This folder contained files for data analysis and figure generation of the Deep Protease Profiling manuscript. It identified significantly enriched peptides, analyzed amino/nucleic acid frequencies, assessed substrate motifs, and deconvoluted protease composition within mixtures. The Python pipeline only included processing from raw fastq files (trimming, filtering, translation) into a merged counts file, with frequency/counts analysis. Subsequent analysis in the manuscript was conducted in R. The full RMarkdown file is available at https://rpubs.com/yue23/protease_analysis
Please specify experiment details/names in parameter.txt, which would include all settings. This pipeline analyzes one-end reads, which could be single or in parallel.
To Start (using Test Files):
- Download this Repository and Extract the Folder
- Run MainFile.py, either by clicking on the Python Script or running the command: python3 MainFile.py
- A Python Popup should appear and the Pipeline would automatically run using the Test Files
- Results would all appear within the file and its folders. This includes filtering, translation, and processing of sequencing data.
- Subsequent analysis was conducted using R, accessed by ProteaseAnalysis.Rmd and PreliminaryAnalysis.Rmd
Prerequisites:
- Python 3 (with Python3 Path setup)
- R
- Installation of other R Packages (specified in the R Scripts)
To Adjust for Different Sequencing Experiments:
- Change Parameters in parameters.txt (# of treatments, # of n_plicates, experiment/treatment names, gzip, filenames, etc.)
- Ensure that the formatting is the same
- Change Seeds (Conserved Sequences) in clean_fastq.py, based on your sequencing
- Each n-mer requires a list of all possible n-mer sequences (e.g. NNK5_combinations.txt). If your n-mer is not 5, please generate your own combinations file using a nested for loop (MakeNNKCombinations.py and move it into the folder. The combinations for 5 is included in the repository.
- Set-Up Sequencing Platform
- Raw FastQ File Processing - Trimming, Filtering, Translation
- Test Files
- R Markdown Files
- Supplementary Data Files
To set up this program, place all the files in the same folder (as organized by default). All folder directories would be set-up automatically, with the program ready to run on the test set. The parameters.txt contains all the editable parameters available. Please edit each while maintaining the same formatting and spacing.
Experiment Criteria: Any number of experiments, with 1 control. The number of n-plicates should be the same for all (e.g. A1, A2, B1, B2, N1, N2, where N is the control). This program accepts one-end reads (single or parallel). If they are paired-end, please merge them prior.
To run the program, place the raw FastQ or Gzip files within the folder and edit the parameters.txt accordingly. Once everything is saved, run MainFile.py
*** Treatment Details ***
Experiment Name = neutrophil_serine_protease
Experiments = cathepsin G, elastase, hPR3, no protease
Control Name = FLAG
Treatments = 15
N-plicates (duplicate/triplicate/etc.) = 3
Treatment Names = C, E, H, V, N
All Treatment Names = C1, C2, C3, E1, E2, E3, H1, H2, H3, V1, V2, V3, N1, N2, N3
*** Mutagenesis Sequencing ***
First Position = 51
Last Position = 65
N-mer = 5
Row Sum Filter = 6
***
Files in Gzip (no/yes) = no
Single/Parallel Read = single
File Names = C1-UDP0001_S1_L001_R1_001.fastq, C2-UDP0002_S2_L001_R1_001.fastq, C3-UDP0003_S3_L001_R1_001.fastq, E1-UDP0004_S4_L001_R1_001.fastq, E2-UDP0005_S5_L001_R1_001.fastq, E3-UDP0006_S6_L001_R1_001.fastq, H1-UDP0007_S7_L001_R1_001.fastq, H2-UDP0008_S8_L001_R1_001.fastq, H3-UDP0009_S9_L001_R1_001.fastq, V1-UDP0013_S13_L001_R1_001.fastq, V2-UDP0014_S14_L001_R1_001.fastq, V3-UDP0015_S15_L001_R1_001.fastq, N1-UDP0010_S10_L001_R1_001.fastq, N2-UDP0011_S11_L001_R1_001.fastq, N3-UDP0012_S12_L001_R1_001.fastq
This program would filter reads where two consecutive reads have a quality score below 28, while only processing reads that contain matched seeds (conserved sequences) that the original genome should contain. This ensures that the reads moving towards DESeq2 were of high quality and reliable.
call(["python3", "clean_fastq.py", name, fileName1, fileName2, gzipChoice, firstPosition, lastPosition])
These reads are then trimmed and translated, based on mutagenesis locations specified. The frequencies of each Amino Acid and Nucleic Acid, at each position, are computed and presented in the files AAFreq and NucFreq. The observed vs expected Amino Acid Frequencies were presented in the files ObsExp. These frequencies were then merged across n-plicates for each experiment/control. Frequencies were visualized using Excel to generate graphs/plots.
call(["python3", "getPeptides.py", name, firstPosition, lastPosition])
call(["python3", "getAminoAcidCount.py", name, firstPosition, lastPosition])
call(["python3", "getNucleicAcidCount.py", name, firstPosition, lastPosition])
call(["python3", "ObservedExpectedPlot.py", name, n_mer])
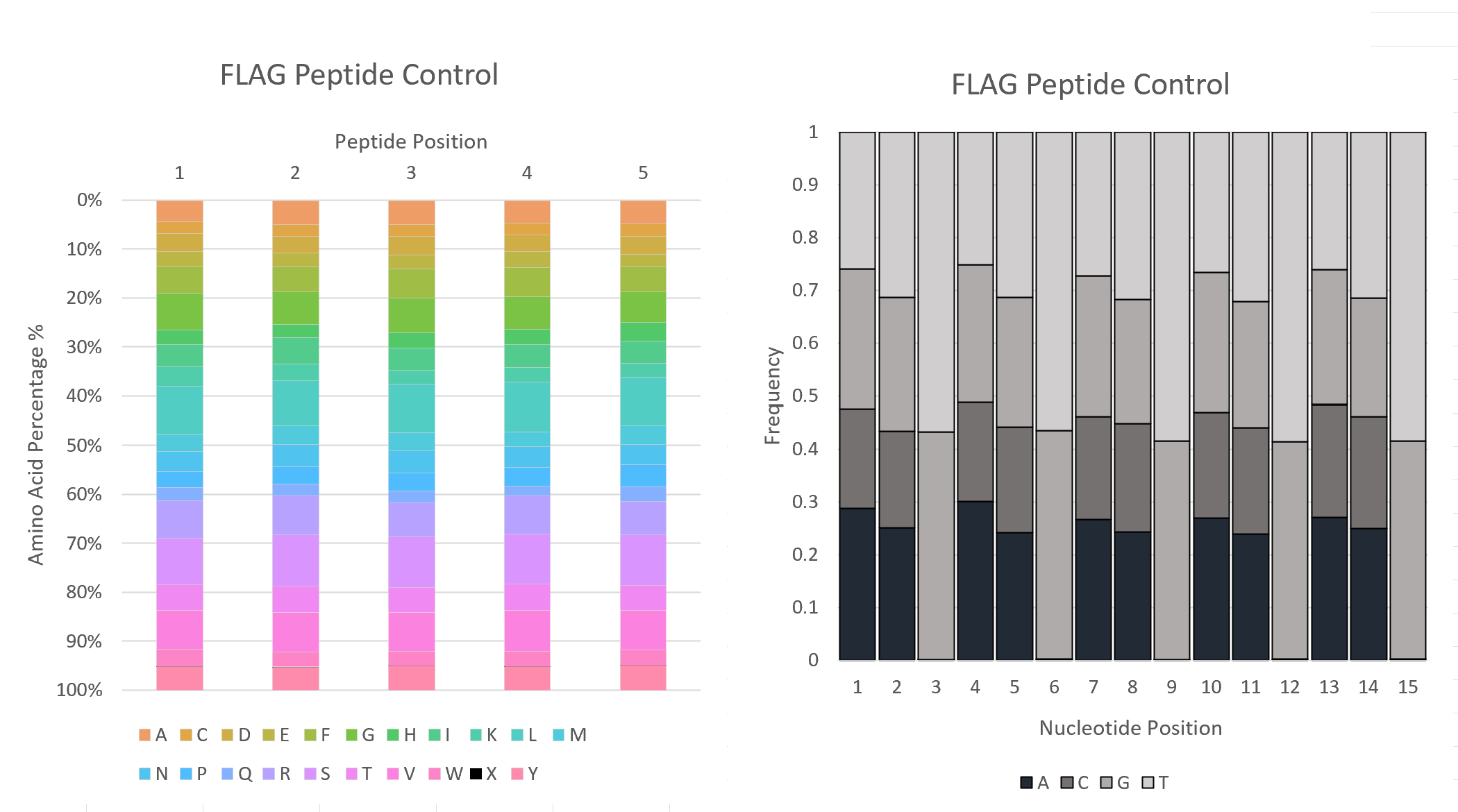
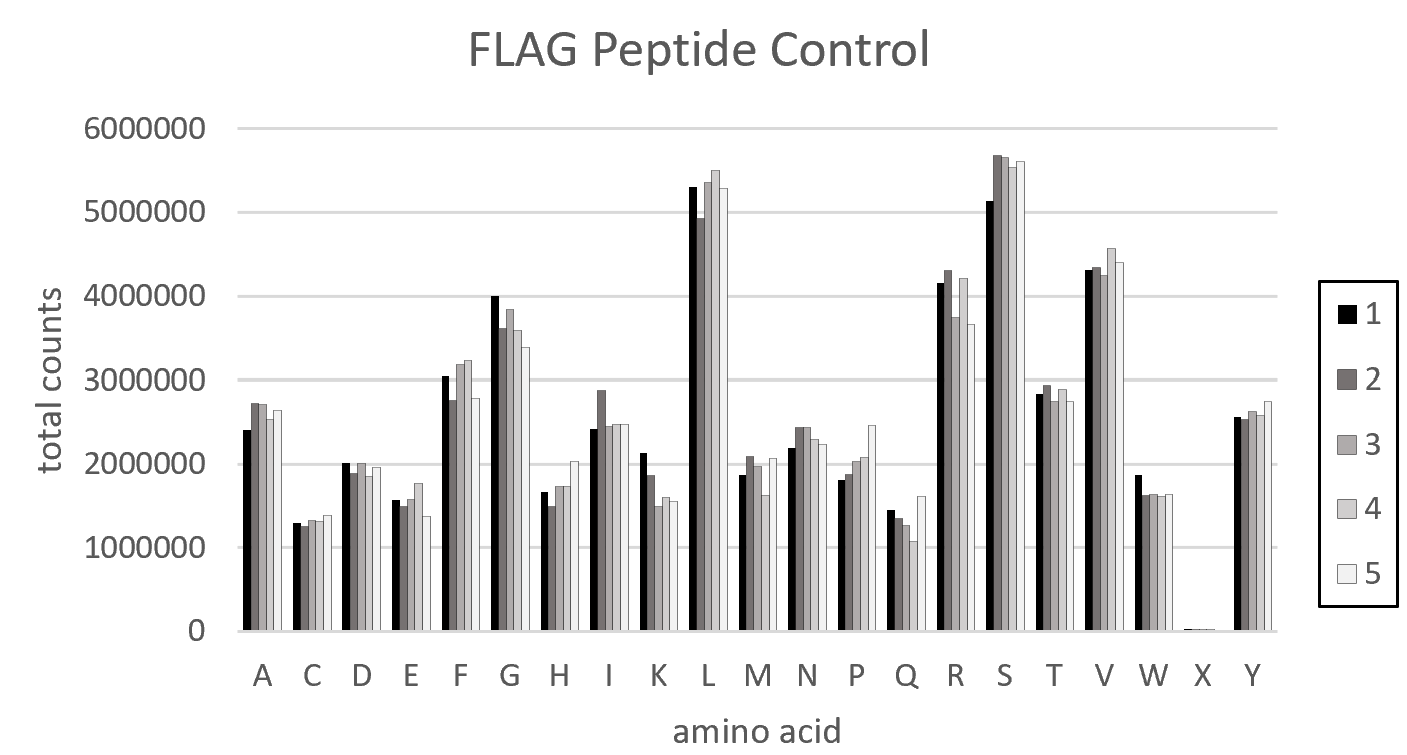
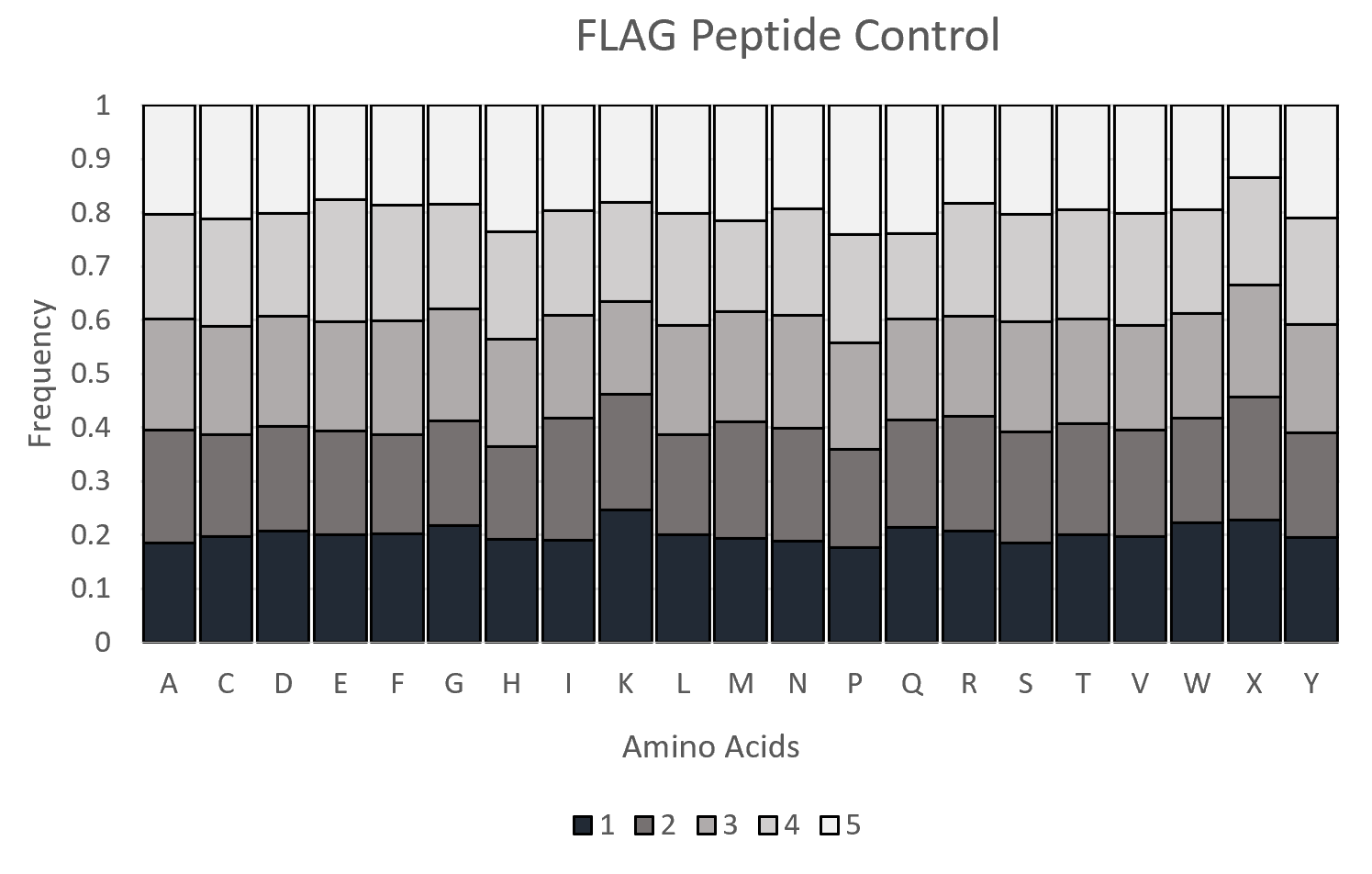
Within the repository (located in the Test Files folder) is a a subset of the FASTQ sequencing results for both neutrophil proteases and supernatant mixtures.
Proteases:
- Cathepsin G: C1-UDP0001_S1_L001_R1_001.fastq, C2-UDP0002_S2_L001_R1_001.fastq, C3-UDP0003_S3_L001_R1_001.fastq
- Elastase: E1-UDP0004_S4_L001_R1_001.fastq, E2-UDP0005_S5_L001_R1_001.fastq, E3-UDP0006_S6_L001_R1_001.fastq
- hPR3: H1-UDP0007_S7_L001_R1_001.fastq, H2-UDP0008_S8_L001_R1_001.fastq, H3-UDP0009_S9_L001_R1_001.fastq
- No Protease: V1-UDP0013_S13_L001_R1_001.fastq, V2-UDP0014_S14_L001_R1_001.fastq, V3-UDP0015_S15_L001_R1_001.fastq
- FLAG Peptide Elution: N1-UDP0010_S10_L001_R1_001.fastq, N2-UDP0011_S11_L001_R1_001.fastq, N3-UDP0012_S12_L001_R1_001.fastq
Activated Supernatant Mixtures:
- w/No Inhibitor: Pool-2-N1_S16_L001_R1_001.fastq, Pool-2-N2_S17_L001_R1_001.fastq, Pool-2-N3_S18_L001_R1_001.fastq
- w/AEBSF: Pool-2-A1_S19_L001_R1_001.fastq, Pool-2-A2_S20_L001_R1_001.fastq, Pool-2-A3_S21_L001_R1_001.fastq
- w/EDTA: Pool-2-E1_S22_L001_R1_001.fastq, Pool-2-E2_S23_L001_R1_001.fastq, Pool-2-E3_S24_L001_R1_001.fastq
- w/AEBSF+EDTA: Pool-2-AE1_S25_L001_R1_001.fastq, Pool-2-AE2_S26_L001_R1_001.fastq, Pool-2-AE3_S27_L001_R1_001.fastq
- FLAG Peptide Elution: Pool-2-G1_S13_L001_R1_001.fastq, Pool-2-G2_S14_L001_R1_001.fastq, Pool-2-G3_S15_L001_R1_001.fastq
The experiments were done in triplicates. The default parameters in parameters.txt were based off this experiment. Parameters for the proteases screen were denoted in 'parameters_protease.txt' and the mixture screen were denoted in 'parameters_mixture.txt'.
All subsequent analyses in the manuscript were conducted in R. All supplemental datasets for the R script were uploaded to the GEO database.
List of Files for running the R script: (place in same folder with R files, located in GEO or GitHub)
- neutrophil_serine_protease_data.csv #counts data of peptides in cleaved/released phages
- neutrophil_mixture_data.csv
- neutrophil_unfiltered_protease&mixture_data.csv
- control_frequency_mixture.txt (sourced from Preliminary Analysis.Rmd) #frequency of amino acids in the unselected phage library
- control_frequency_protease.txt
- control_frequency_combined.txt
- cathepsin G_MEROPs.xlsx (sourced from MEROPs substrate lists online) #8-mer substrates reported on MEROPs for each protease
- elastase_MEROPs.xlsx
- hPR3_MEROPs.xlsx
- aligned_sig_peptides_cathepsin G.fasta (generated from code in R file) #aligned peptides but prior to centering at P1
- aligned_sig_peptides_elastase.fasta
- aligned_sig_peptides_no protease.fasta
- aligned_sig_peptides_hpr3.fasta
- aligned_sig_peptides_no inhibitor.fasta
- aligned_sig_peptides_AEBSF.fasta
- aligned_sig_peptides_EDTA.fasta
- aligned_sig_peptides_AEBSF+EDTA.fasta
List of Files for running Icelogo (sourced from Preliminary Analysis.Rmd) #peptide lists that capture the AA frequency in the unselected phage library
- control_frequency_protease.fasta
- control_frequency_mixture.fasta
- control_frequency_protease_9mer.fasta
- control_frequency_mixture_9mer.fasta
- control_frequency_combined.fasta
The complete high-throughput sequencing reads of cleaved/release phage DNA from Nextseq2000 were uploaded onto SRA (Accession ID: PRJNA1141700). Filtered and unfiltered counts tables of cleaved peptides were uploaded onto GEO.
Other data files uploaded to GitHub:
- Peptides determined to be significantly cleaved (unaligned and aligned)
- Positional weight matrices of each condition (unaligned and aligned)
- Synthetically aligned peptides
- Frequencies the Unselected Phage Library
- MEROPs substrates of the 3 neutrophil serine proteases
The code adapts work from:
- Dr. Kart Tomberg https://github.com/tombergk/NNK_VWF73/
- Dr. Matt Holding https://github.com/Matthew-Holding
mod575.py: written by Dr. Kart Tomberg
Substrate Analysis using Alphafold2: from Dr. Matt Holding
Blood Protease Dataset: Kretz CA, Tomberg K, Van Esbroeck A, Yee A, Ginsburg D. High throughput protease profiling comprehensively defines active site specificity for thrombin and ADAMTS13. Sci Rep. 2018 Feb 12;8(1):2788.
DESEQ2: Love MI, Huber W, Anders S (2014). “Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2.” Genome Biology, 15, 550. https://doi.org/10.1186/s13059-014-0550-8.
MClust: Scrucca L, Fop M, Murphy TB, Raftery AE (2016). “mclust 5: clustering, classification and density estimation using Gaussian finite mixture models.” The R Journal, 8(1), 289–317. https://doi.org/10.32614/RJ-2016-021.
CibersortX: Newman AM, Steen CB, Liu CL, Gentles AJ, Chaudhuri AA, Scherer F, et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat Biotechnol. 2019 Jul;37(7):773–82.
EPIC: Racle J, Gfeller D. EPIC: A Tool to Estimate the Proportions of Different Cell Types from Bulk Gene Expression Data. Methods Mol Biol. 2020;2120:233–48.
DECIPHER: Wright ES. DECIPHER: harnessing local sequence context to improve protein multiple sequence alignment. BMC Bioinformatics. 2015 Oct 6;16(1):322.
Other Packages Used: dplyr, tidyr, RColorBrewer, reshape, factoextra, stringr, openxlsx, stats, multcomp, Hmisc, gridExtra, tidyverse, ggplot2, ggseqlogo, ggpubr, ggbeeswarm, ggVennDiagram, ggVenn, pheatmap, PoiClaClu, BiocManager, S4Arrays, DelayedArray, plotly, plot3D, dbscan, forcats, umap, immunedeconv, plotly, clusterSim