This folder contains files involved in the high throughput sequencing analysis from the 5 Amino Acid Mutagenesis sequencing data. It identifies significantly enriched peptides, analyzes the amino/nucleic acid frequencies, and clusters the enriched peptides based on their properties (Principal Component Analysis). The pipeline is all outlined in order within NmerGene.py, which automatically calls the rest of the scripts as long as they are in the same folder.
This program can accommodate any n-mer mutagenesis library at any location, for any number of experments/replicates. Please specify these details in parameter.txt, which has all the default settings. It analyzes one-end reads, which could be single or in parallel.
To Start (based on default parameters, testing data):
- Download this Repository and Extract the Folder
- Run NmerGene.py, either by clicking on the Python Script or running the command: python3 NmerGene.py
- A Python Popup should appear and the Pipeline would automatically run using the Test Files
- The Plots and Reults would all appear within the file and its folders
Prerequisites:
- Python 3 (with Python3 Path setup)
- R (with R Script Path setup)
- DESeq2
- MClust
- Weblogo (with Ghostscript installed)
- Installation of Other R Packages for Visualization (specified in the R Scripts)
To Adjust for different Sequencing Experiments:
- Change Parameters in parameters.txt (# of treatments, # of n_plicates, experiment/treatment names, gzip, etc.)
- Ensure that the formatting is the same
- Change Seeds (Conserved Sequences) in clean_fastq.py, based on your sequencing
- Each n-mer requires a list of all possible n-mer sequences (e.g. NNK5_combinations.txt). If your n-mer is 6 or above, please generate your own combinations file using a nested for loop (MakeNNKCombinations.py and move it into the folder. The combinations for 5 and below are included in the repository (filesize too large for >5 n-mers).
- Set-Up NmerGene
- Raw FastQ File Processing - Trimming, Filtering, Translation
- DESEQ2 Analysis for Significantly Enriched Peptides using R
- Principal Component Analysis to cluster Significatly Enriched Peptides
- Weblogo Analysis for clusters
- Test Files
To set up this program, place all the files in the same folder (as organized by default). All folder directories would be set-up automatically, with the program ready to run on the test set (run NmerGene.py). The parameters.txt contains all the editable parameters available. Please edit each while maintaining the same formatting and spacing.
Experiment Criteria: Any number of experiments, with 1 control. The number of n-plicates should be the same for all (e.g. A1, A2, B1, B2, N1, N2, where N is the control). This program accepts one-end reads (single or parallel). If they are paired-end, please merge them prior.
To run the program, place the raw FastQ or Gzip files within the folder and edit the parameters.txt accordingly (below). Once everything is saved, run NmerGene.py
*** Treatment Details ***
Experiment Name = NeutrophilProteases
Experiments = cathepsinG, elastase, hpr3
Control Name = N
Treatments = 8
N-plicates (duplicate/triplicate/etc.) = 2
Treatment Names = C, E, H, N
All Treatment Names = C1, C2, E1, E2, H1, H2, N1, N2
*** Mutagenesis Sequencing ***
First Position = 55
Last Position = 69
N-mer = 5
Min Count for each peptide (for DESEQ2) = 6
***
Files in Gzip (no/yes) = no
Single/Parallel One-End Read = parallel
*** Principal Component Analysis ***
Principal Components Used = 3
Limit of Clusters = 10
This program would filter reads where two consecutive reads have a quality score below 28, while only processing reads that contain matched seeds (conserved sequences) that the original genome should contain. This ensures that the reads moving towards DESEQ2 are of high quality and reliable.
call(["python3", "clean_fastq.py", name, fileName1, fileName2, gzipChoice, firstPosition, lastPosition])
These reads are then trimmd and translated, based on the mutagenesis locations specified. The frequencies of each Amino Acid and Nucleic Acid, at each position, are computed and presented in the files AAFreq and NucFreq. The observed vs expected Amino Acid Frequencies are presented in the files ObsExp. These frequencies are then merged across n-plicates for each experiment/control. Further analysis can be conducted on the frequencies to generate graphs/plots.
call(["python3", "getPeptides.py", name, firstPosition, lastPosition])
call(["python3", "getAminoAcidCount.py", name, firstPosition, lastPosition])
call(["python3", "getNucleicAcidCount.py", name, firstPosition, lastPosition])
call(["python3", "ObservedExpectedPlot.py", name, n_mer])
R conducts differential expression analysis using DESEQ2 to identify significantly enriched peptides. This program uses the Bonferroni Correction and defines enriched peptides as those with a p value less than 0.05 and a log fold change greater than 0.
The generated DESEQ results are then converted into FASTA files for PCA Analysis.
subprocess.call(["Rscript", os.getcwd() + "/DESEQprocessor.R", os.getcwd(), colData, replicates, experimentData, control[0], experimentName], shell=True)
for i in range(len(experiments)):
fileName1 = find_files('enrich_'+experiments[i]+'_bonf_deseq2.txt')
call(["python3", "DESEQtoFASTA.py", experiments[i], fileName1])
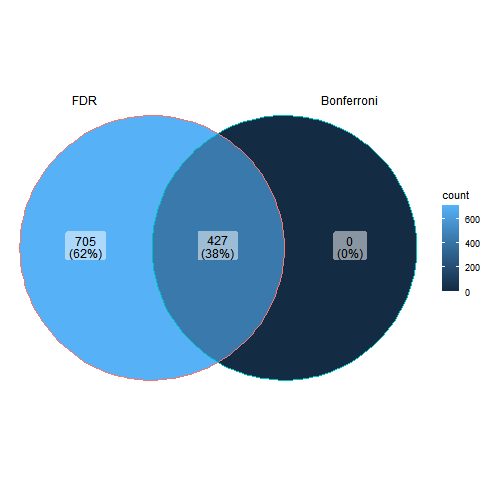
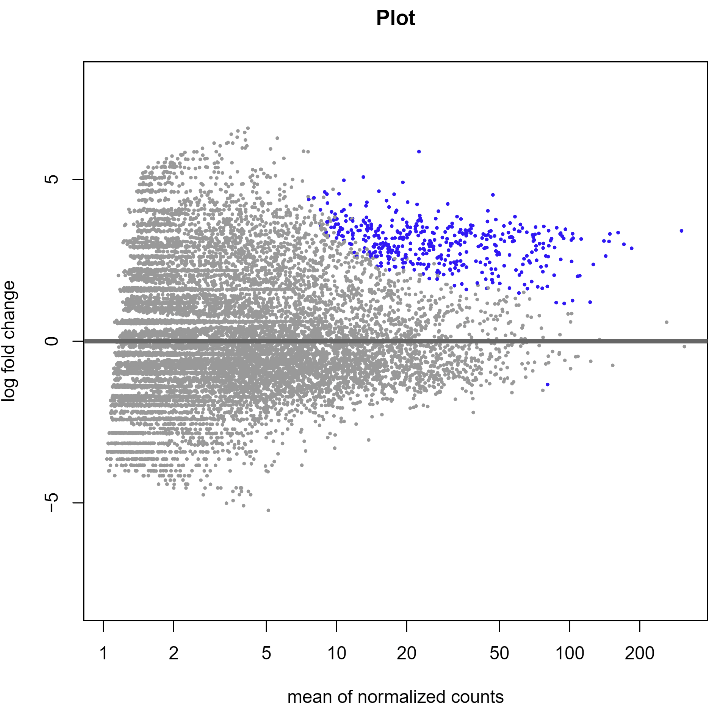
DESEQ2 Results for Cathepsin G on the Test Files Subset
Principal Component Analysis uses key properties of Amino Acids (Charge, Disorder, HPATH, RMW) and attaches a weight to each position's Amino Acid properties. This creates a principal component. PCA Analysis aims to create principal components in a way that generates the furthest separation between groups, forming clusters of peptides with similar properties. This is crucial towards deconvoluting the peptides and identifying peptide signatures that reveal information about enrichment.
The default is 3 Principal Components and 10 Maximum Cluters. These can be adjusted through parameters.txt
subprocess.call(["Rscript", os.getcwd() + "/PCAprocessor.r", os.getcwd(), experimentData, PCAcomponents, PCAclusters], shell=True)
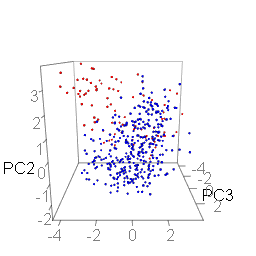
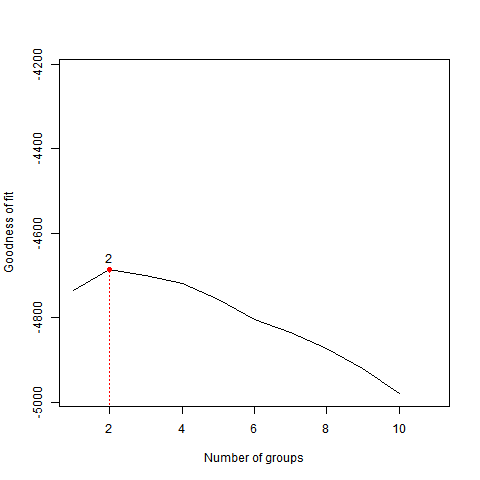
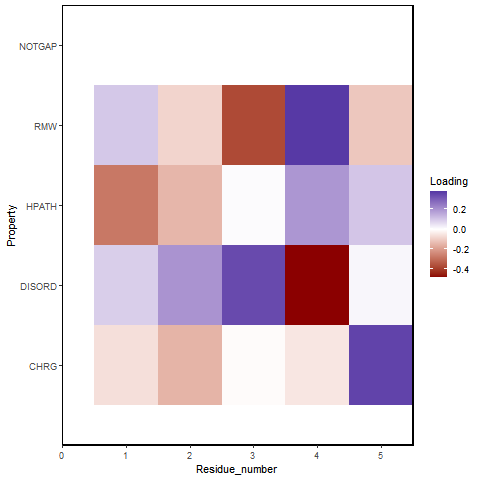
PCA Results for Cathepsin G on the Test Files Subset
To visualize the peptide motifs of each cluster, sequence logos are created using Weblogo software.
call(["python3", "WeblogoProcessor.py", treatmentList])
![]()
![]()
Weblogos for Cathepsin G on the Test Files Subset
Within the repository (located in the Test Files folder) is a a subset of the FASTQ sequencing results for Neutrophil Proteases Cathepsin G (C), Elastase (E), and Human Proteinase 3 (H) with an unselected control (N). The experiments were done in duplicates. The default parameters in parameters.txt were based off this experiment.
The code adapts work from:
- Dr. Kart Tomberg https://github.com/tombergk/NNK_VWF73/
- Dr. Matt Holding https://github.com/Matthew-Holding
mod575.py: written by Dr. Kart Tomberg
PCA Analysis Files: from Dr. Matt Holding
DESEQ2: Love MI, Huber W, Anders S (2014). “Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2.” Genome Biology, 15, 550. https://doi.org/10.1186/s13059-014-0550-8.
MClust: Scrucca L, Fop M, Murphy TB, Raftery AE (2016). “mclust 5: clustering, classification and density estimation using Gaussian finite mixture models.” The R Journal, 8(1), 289–317. https://doi.org/10.32614/RJ-2016-021.